
Welcome!
Welcome to the new CancerGRACE.org! Explore our fresh look and improved features—take a quick tour to see what’s new.
Introduction to Molecular Markers
A molecular marker is an identifiable molecular characteristic (usually DNA, RNA, or protein) in a patient or a tumor that can be used to provide prognostic or predictive information about the cancer or about a particular treatment. A prognostic marker is one which indicates a better or worse outcome irrespective of treatment. For example, a mutation in the KRAS gene has been widely regarded as a poor prognostic molecular marker (see below), but does not necessarily guide us in selecting therapy for a particular patient. In contrast a predictive marker indicates a better or worse chance of an outcome for a specific treatment. Identifying this type of marker is a major goal of translational research and forms the basis of "personalized medicine", which is simply saying that you may be able to determine ahead of time which treatment will or will not work in a specific patient.
In this chapter, I will try and describe the most common molecular markers being investigated in lung cancer, including some tests that are already being used in practice today.
The Epidermal Growth Factor Receptor (EGFR)
EGFR Mutations
The first real evidence that a molecular marker could predict treatment outcome in NSCLC came about in the early 2000s with the targeted drugs that inhibited the epidermal growth factor receptor (EGFR). The EGFR tyrosine kinase inhibitors (TKIs) Iressa (gefitinib) and Tarceva (erlotinib) were being tested in large numbers of patients with advanced non-small cell lung cancer. EGFR was thought to be an important target in lung cancer because most NSCLC tumors express it, and high levels of expression were associated with a worse prognosis. In a phase III ISEL trial, Iressa did not improve overall survival compared to placebo treatment in previously treated NSCLC patients, but the similar BR.21 trial (testing Tarceva rather than Iressa) did show a modest (~2 month) improvement in overall survival in previously treated NSCLC patients. This led to the approval of Tarceva in all NSCLC patients who had failed one or two prior chemotherapy regimens.
However, what was immediately evident from these trials was that about 10% of Western patients treated with either of these drugs had dramatic and sometimes long-lasting responses. When they looked at the responders, they found that a large proportion were women, all had adenocarcinoma, many were of Asian ethnicity, and most had either never smoked or smoked very little compared to average NSCLC patients (http://cancergrace.org/lung/2006/11/18/is-tarceva-only-useful-in-certain-patient-groups/). In 2004, investigators at the Dana Farber Cancer Institute (http://www.sciencemag.org/cgi/content/full/304/5676/1497) and at Massachusetts General Hospital (http://www.nejm.org/doi/full/10.1056/NEJMoa040938) in Boston, and also at Memorial Sloan Kettering Cancer Center in NYC (http://www.pnas.org/content/101/36/13306.long) , simultaneously published results showing that most of these "dramatic responders" had recurring mutations in the tyrosine kinase (TK) domain of the EGFR gene.
The EGFR protein sits in the cell membrane and straddles the inside and outside of the cell.
 (click on image to enlarge)
(click on image to enlarge)
Sequist el al., JCO 2007
The TK is the part of the protein, located inside the cell, which "switches on" when a growth factor (or ligand) from outside the cell binds to the outside portion of the EGFR. This switch, when flipped on, allows the EGFR to signal the cell to grow and survive. In the NSCLC patients who have mutations in the TK domain of the EGFR, very little growth factor is needed to flip on the switch, and once turned on the cancer cell is driven to grow and divide essentially through this one signal. This makes the cancer cell exquisitely sensitive to dying when the switch is turned off by a drug like Iressa or Tarceva, and explains why some patients can do so well on these drugs.
The EGFR gene has 28 exons (parts of the DNA that serve as the blueprint for the EGFR protein), and exons 18 through 21 code for the TK part of the receptor. Although there can be mutations anywhere in the TK domain, only some of them confer sensitivity to the TKIs.

Sharma et al., Nature Reviews Cancer 2007
About 45% of sensitizing mutations are what are called in frame deletions in exon 19, making them the most common EGFR mutations. Deletion mutations result when short segments of the DNA are removed (deleted) from the DNA, but without interrupting the blueprint. About 40-45% of the sensitizing mutations are point mutations in exon 21, the most common being L858R (At the "point" in the 858th position, the normal amino acid leucine (L) is switched out for an arginine (R), which changes the protein function). A number of prospective trials have now shown that the presence of an activating EGFR mutation in the tumor predicts a high likelihood of objective tumor response, up to 70-80%. These trials will be described in more detail in another chapter.
Most of the remaining mutations don't cause the EGFR to be sensitive to EGFR TKIs (I.e are resistant to the TKIs), but importantly some of them do still turn on the EGFR and drive the cancer cell to grow. The most important of these is the T790M, a point mutation in exon 20 resulting in the substitution of methionine (M) for threonine (T). This mutation seems to allow the EGFR TK to work much better than normal, so that the TKI no longer has an advantage in binding over the normal ligand, something called ATP. Mutations (usually in this case called insertion mutations) in exon 20 have also been associated with resistance.
Mutations can be detected using a number of methods, although only two are in commercial use at this time. Direct sequencing of exons 18 through 21 of the EGFR gene is currently done by a company called Genzyme (Cambridge, MA), and can be performed on either paraffin-embedded or fresh-frozen tumor samples. In DNA sequencing, the actual nucleotides that make up the DNA blueprint are individually identified and placed in the correct order, allowing us to identify any mistakes (mutations). Sequencing is the only method that can identify every mutation in the tyrosine kinase domain, whether predictive of responsiveness to TKIs or not.
The second commercially available method uses something called allele-specific polymerase chain reaction (PCR) technology to specifically amplify (make lots of copies of) sections of the EGFR gene with common mutations, which can then be detected by a machine. One example of allele-specific amplification is known as Scorpion ARMS (Amplification Refractory Mutation System), developed by a company called DxS. This method only detects 28 of the most common EGFR mutations, but generally requires smaller amounts of tissue than sequencing and has a slightly faster turnaround time. There is also evidence that this method may be more sensitive than direct sequencing, leading to fewer false negative tests.
EGFR Overexpression by Immunohistochemistry (IHC) or fluorescence in situ hybridization (FISH)
EGFR protein overexpression, which can be detected by using IHC to stain the tumor for the protein, is very common in NSCLC and has been reported to be a poor prognostic marker. However, protein expression has not proven to be an important marker predicting benefit from anti-EGFR therapy.
One other potential marker of benefit that has been extensively tested is EGFR gene amplification by fluorescence in situ hybridization (FISH; see figure). This composite marker is termed FISH-positive and includes both true gene amplification (tight gene clusters with a gene-to-chromosome ratio per cell of ≥ 2, or ≥ 15 copies of EGFR per cell in ≥ 10% of cells analyzed) and chromosome polysomy (≥ 4 copies in ≥ 40% of cells).

Cappuzzo, JNCI 97:643, 2005.
The red dots are fluorescently-labeled copies of the EGFR gene. Figure A shows the normal 2 copies of EGFR per cell. C shows multiple chromosomes per cell (high polysomy), and D shows increased copies of the EGFR gene, both of which are considered FISH-positive.
Interestingly, it appears that the majority of patients with EGFR mutations are also FISH-positive, making interpretation of these individual tests as predictive markers more complicated. Although there was early evidence that EGFR FISH-positivity was a predictive marker for benefit from both EGFR TKIs and anti-EGFR antibodies such as Erbitux (cetuximab) recent evidence suggests that this is probably not the case. This evidence will be described in more detail in another chapter.
Serum Proteomic Profiling
An intriguing new potential molecular marker is something called a serum proteomic profile, which gives a snapshot of many different proteins in the blood at one time. This is appealing for a number of reasons, not the least of which is that it does not require a biopsy of the tumor. Proteomic profiling in lung cancer has been pioneered by Dr. David Carbone at Vanderbilt University, and utilizes a technology called MALDI (Matrix-assisted laser desorption/ionization mass spectrometry) to identify the serum proteins.

Yildiz et al., JTO 2008.
Proteomic profile for both normal controls and patients with NSCLC.
There is evidence that proteomic profiles from the blood of NSCLC patients can predict which patients are more or less likely to benefit from EGFR TKIs such as Iressa or Tarceva. These profiles have been used to create a commercially available test called Veristrat (Biodesix), which appears to be able to stratify patients who received EGFR TKIs into good or poor prognostic groups in retrospective analyses and might be useful in determining which patients are unlikely to benefit from such drugs. To date, however, this test had not been validated in a prospective clinical trial, and so should be considered experimental until this occurs. Prospective validation is underway in a European trial called the PROSE study.
KRAS Mutations
The Kirsten rat sarcoma virus (K-RAS) gene codes for a very important protein located at the plasma membrane called a "g-protein", which serves as an intermediary switch between receptors at the membrane and intracellular signals which regulate cell growth and survival. Although normal RAS proteins cause no problems, if they are mutated they can lead to the development of cancer by constantly signaling the cell to grow and survive even when it should not. This is known as oncogenesis and mutant KRAS, like mutant EGFR, is an oncogene.
KRAS mutations are present in up to 30% of all human cancers, and in particular are present in about 20% of adenocarcinomas of the lung. KRAS mutations are strongly associated with tobacco smoke, and are also present in the vast majority of a subtype of NSCLC known as mucinous bronchioloalveolar carcinoma (BAC). Interestingly, KRAS and EGFR mutations appear to be mutually exclusive. Since KRAS is "downstream" of the EGFR, mutant KRAS takes EGFR out of the loop. Think of EGFR mutation as a light switch that is always on, while KRAS bypasses the wiring inside the wall so that the lights are always on, a different means to the same end.

Studies have convincingly shown that the presence of a KRAS mutation is a poor prognostic marker for survival in NSCLC. There is also evidence that patients with KRAS mutations may not benefit from adjuvant chemotherapy, and that these tumors tend not to respond to the EGFR TKIs.
What is less obvious is whether KRAS mutations are predictive of a complete lack of benefit from EGFR TKIs, or if they simply have the same low objective response rate seen in all EGFR wild-type tumors. This will be detailed more in a separate chapter on utilizing molecular markers for therapy, but for now KRAS mutations should not preclude patients from receiving EGFR TKIs in the second and third-line treatment of advanced NSCLC.
KRAS mutations can be detected using the same type of tests used to detect EGFR mutations, namely sequencing or PCR-based assays, and this testing is routinely available in many hospitals.
EML4-ALK translocations
One of the most intriguing new molecular markers identified in patients with NSCLC in recent years is the EML4-ALK fusion gene, resulting from an inversion in chromosome 2 that juxtaposes the EML4 gene with the ALK gene. Since its first description, this gene translocation has been identified in approximately 4% of patients with NSCLC, most commonly in younger males, never smokers, and patients with adenocarcinoma. This new oncogene also appears to be mutually exclusive of EGFR and KRAS mutations. The EML4-ALK translocation is a very exciting new target and potential predictive marker for benefit from drugs that inhibit ALK, but these compounds are still in early clinical development and are only available as part of a clinical trial. This will be discussed further in a separate chapter.
The EML4-ALK translocation can be easily visualized on paraffin embedded tumor tissue using a technology called break-apart fluorescence in situ hybridization (FISH; see Figure). The red and green fluorescent probes label the ALK gene in different areas on either side of the breakpoint. In a normal cell, the red and green are right next to each other on chromosome 2, but when the ALK translocation is present the probes separate and are farther away from each other (see figure below). This is a subtle change and requires expert interpretation to avoid false positive or negative results.

Bang et al., ASCO 2010
There is evidence that most patients with ALK translocations also have overexpression of the fusion protein, which can be detected by the technically simpler technique of IHC staining. However, this has not yet been completely validated and FISH remains the gold standard for now.
Markers of DNA Repair (ERCC1, MSH2, and RRM1)
Although we spend a great deal of time in the academic lung cancer world talking about novel agents that inhibit EGFR and ALK, the reality is that traditional cytotoxic chemotherapy remains the primary treatment for the majority of advanced NSCLC patients. Since any particular chemotherapy treatment only causes clinical benefit in a subset of patients, molecular markers that predict potential benefit from existing chemotherapy drugs have the potential to help more NSCLC patients than the sexiest of the new molecular markers detailed above.
The backbone of chemotherapy treatment for NSCLC is platinum (cisplatin and carboplatin), usually used in combination with one of a number of other chemotherapy agents as a platinum-doublet. Platinum works by binding to cellular DNA and creating adducts (essentially causing the DNA to stick together so that it cannot replicate the way it should). These adducts are sensed by the cellular DNA repair machinery and the DNA is either repaired or causes the cell to die through a process known as apoptosis. One of the most important proteins in the repair of platinum-DNA adducts is excision repair cross complementation group-1 (ERCC1). Cancer cells with high levels of ERCC1 protein are better able to repair the DNA adducts before the cell can die, and thus are resistant to platinum compounds. In contrast, NSCLC cells with low levels of ERCC1 have a worse prognosis but are more sensitive to platinum chemotherapy. This finding has led to a number of clinical trials that attempt to determine ERCC1 levels ahead of time and use the platinum only when the levels are low. These trials in advanced NSCLC will be explored more in a separate chapter, and were reviewed in early stage NSCLC by Dr. Wakelee.
Another DNA repair protein involved in platinum sensitivity and resistance is known as MutS homolog 2 (MSH2). Similar to ERCC1, low levels may predict sensitivity to platinum and high levels resistance. More importantly, the combination of ERCC1 and MSH2 levels may be a better predictor of platinum sensitivity than either alone. The MSH2 story is still relatively new and its role as a molecular marker has not been well tested to date.
A third molecular marker of chemosensitivity that has been extensively tested is ribonucleotide reductase-M1 (RRM1). RRM1 is the regulatory subunit of an important enzyme (ribonucleotide reductase (RR)) that is involved in production of the building blocks of DNA, and RR is inhibited by the chemotherapy drug gemcitabine. When RRM1 levels are low, cancer cells are more sensitive to gemcitabine, while high RRM1 levels may predict resistance to the drug.
Expression levels of ERCC1, MSH2, and RRM1 can be assessed by two methods: 1) staining for protein using IHC, and 2) assessing RNA levels using quantitative RT-PCR technology. Testing RNA levels is much more technically difficult than IHC staining, and usually requires fresh frozen tumor tissue for the test. IHC can be done on archived, paraffin embedded tumor tissue (see below).

Olaussen et al,. NEJM 355;10
Conclusion
Molecular markers are identifiable signals in patients or in tumor tissue that have the potential to provide information about prognosis or even to predict which treatments work best in an individual cancer patient. They form the backbone of personalized medicine, which in the not-too-distant future will be how all NSCLC patients are treated. In some ways, such as testing for EGFR mutations to guide the use of EGFR TKIs, we are already taking small steps into this exciting new arena. However, much work still needs to be done before every lung cancer patient can be matched with the most effective and least toxic treatment.
We thank Pfizer Oncology for the educational grant that made the Lung Cancer Reference Library possible.
Please feel free to offer comments and raise questions in our
discussion forums.



Hi app.92, Welcome to Grace. I'm sorry this is late getting to you. And more sorry your mum is going through this. It's possible this isn't a pancoast tumor even though...
A Brief Tornado. I love the analogy Dr. Antonoff gave us to describe her presentation. I felt it earlier too and am looking forward to going back for deeper dive.
Dr. Singhi's reprise on appropriate treatment, "Right patient, right time, right team".
While Dr. Ryckman described radiation oncology as "the perfect blend of nerd skills and empathy".
I hope any...
My understanding of ADCs is very basic. I plan to study Dr. Rous’ discussion to broaden that understanding.
Here's the webinar on YouTube. It begins with the agenda. Note the link is a playlist, which will be populated with shorts from the webinar on specific topics
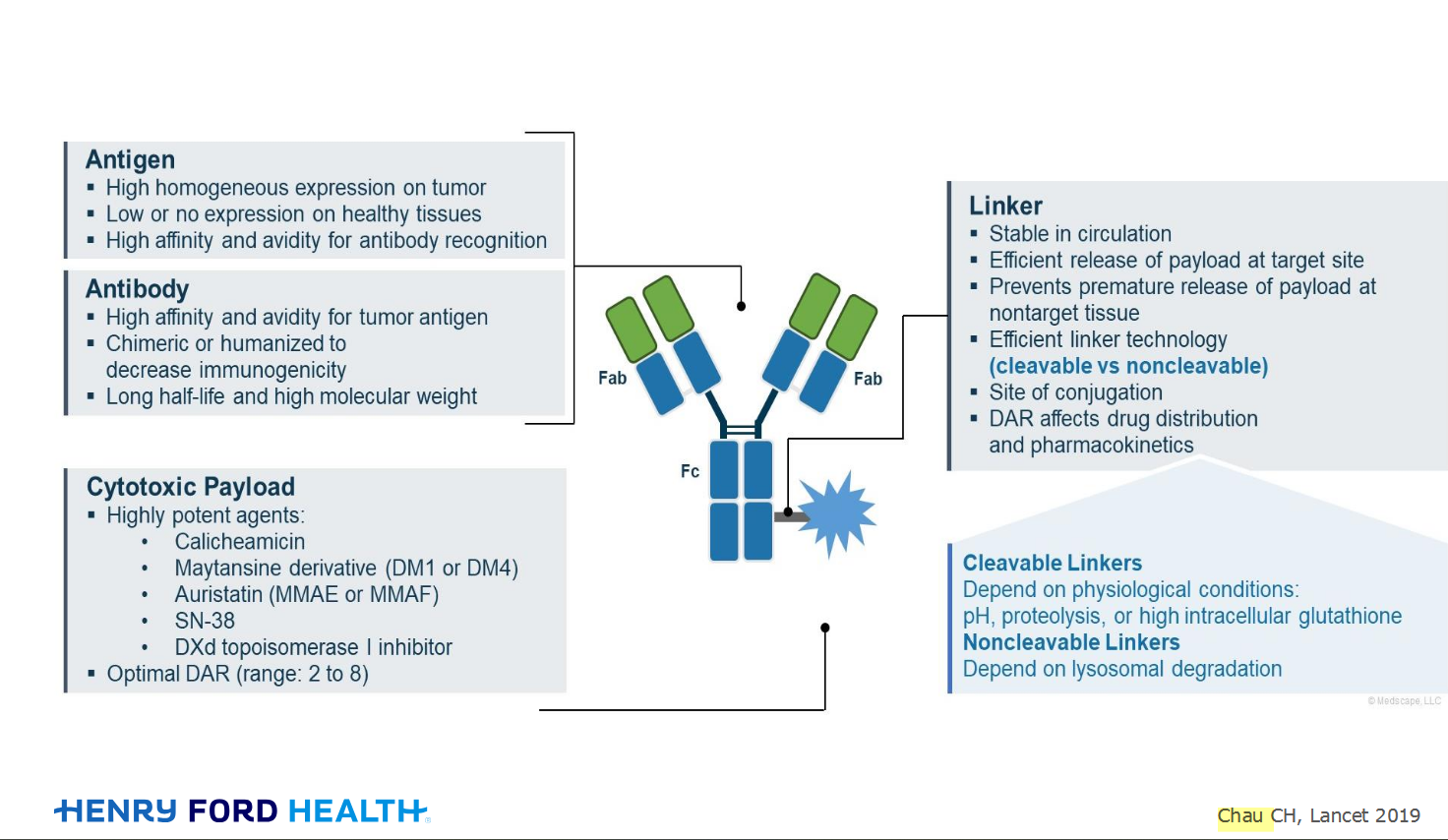
An antibody–drug conjugate (ADC) works a bit like a Trojan horse. It has three main components:

Welcome to the new CancerGRACE.org! Explore our fresh look and improved features—take a quick tour to see what’s new.